
Microscopia de fluorescência
Foram assinalados vários problemas nesta página ou se(c)ção: |

O microscópio de fluorescência é uma variação do microscópio de luz ultravioleta, que permite a observação da fluorescência de objetos, através da emissão de raios de um determinado comprimento de onda. Esta tecnologia contribuiu significativamente para o estudo da biologia celular. A imagem é produzida pela radiação electromagnética emitida pelas moléculas no estado excitado, que possui maior comprimento de onda.
Em geral, a fluorescência é a propriedade que algumas substâncias têm de emitir radiação com maior comprimento de onda, após serem excitadas com radiação de baixo comprimento de onda. Deste modo, certas substâncias, depois de absorverem luz ultravioleta, emitem radiação no espectro visível. Para deixar passar somente a emissão secundária desejada, devem-se colocar filtros apropriados debaixo do condensador e por cima das objetivas.
Este microscópio é geralmente usado para detectar substâncias com autofluorescência, como por exemplo a vitamina A; ou substâncias marcadas com fluorocromos, que poderão ser conjugados com anticorpos para localizar e identificar moléculas individuais que reagem com o anticorpo. Esta aplicação é designada por imunofluorescência, sendo uma das mais importantes no contexto da microscopia de fluorescência.
História
[editar | editar código-fonte]Fluorescência [1]
[editar | editar código-fonte]A sua descoberta é atribuída a Nicolás Monardes (1577), um médico e botânico espanhol. A descrição da opalescência azul surgiu a partir de uma infusão de água e de madeira de uma pequena árvore mexicana. No entanto, foi Bernardino de Sahagún, um missionário franciscano, que descreveu esta propriedade da madeira (já antes observada pelos astecas).
Esta madeira (Lignum nephriticum) foi muito popular na Europa dos séculos XVI e XVII, devido às suas propriedades medicinais no tratamento das maleitas renais. O inglês John Frampton também a descreveu como sendo “uma madeira branca que dá uma cor azul” quando colocada em água.
Em 1646, o padre jesuíta alemão Athanasius Kircher escreveu um livro no qual descreveu a sua observação sobre este fenômeno. Segundo ele, a luz que atravessava a infusão aquosa desta madeira apresentava uma cor amarelada, enquanto a luz refletida por esta solução apresentava uma cor azul.
Em 1670, Robert Boyle investigou este fenômeno mais profundamente, descobrindo que após muitas infusões a madeira perdia a sua capacidade de conferir uma cor à água. Concluiu, desta forma, que havia algum sal essencial na madeira responsável por este efeito, e que a adição de um ácido abolia a cor, enquanto a adição de uma base permitia restaurá-la. Boyle foi, assim, o primeiro a utilizar a fluorescência como indicador de pH.
Apesar de nos séculos seguintes não ter sido muito estudada, em 1915 Safford identificou as espécies que produziam esta madeira como Eynsemhardtia polystachia. Mais recentemente, foram isolados alguns glucosilhidroxialcanos altamente fluorescentes a partir desta planta.
Outros estudos recentes, de Ulises Acuna, indicaram que a emissão azul original observada pelos astecas era provavelmente devida à conversão da coatline B, sob condições ligeiramente alcalinas, num composto com forte emissão de radiação azul (com pico máximo de emissão a 466 nm), que fazia lembrar a fluoresceína.
Ainda no século XVII, em 1612, Galileu Galilei descreveu a emissão de luz a partir de uma pedra famosa, a “Bolognian stone” – fosforescência.
Em 1833, David Brewster descreveu que quando um feixe de luz branca passa através de uma solução alcoólica de folhas é observado um feixe de luz vermelha – fluorescência da clorofila.
Doze anos depois, em 1845, John Herschel observou pela primeira vez a fluorescência a partir do sulfato de quinina, designando este fenómeno de dispersão epipólica. Em 1852, George Gabriel Stokes denominou este mesmo fenómeno de reflexão dispersiva. Na sua experiência, Stokes usou um prisma para dispersão do espectro da luz solar e iluminação de uma solução de quinina. Este verificou que só obtinha algum efeito quando a solução era colocada na região ultravioleta do espectro. Desta forma, a fluorescência apresenta um comprimento de onda superior ao da fonte de excitação (luz ultravioleta) – “desvio de Stokes”.
Em 1882, Paul Erlich usou a uranina (o sal sódico da fluoresceína) para observar a secreção do humor aquoso no olho. Esta foi a primeira utilização in vivo da fluorescência.
Em 1887, K. Noack publicou um livro no qual listava 660 compostos organizados de acordo com a cor da sua fluorescência.
Dez anos depois, R. Meyer utilizou o termo “fluoróforo” para descrever os grupos químicos que se encontravam associados à fluorescência. Este termo é análogo ao termo “cromóforo”, o qual foi usado pela primeira vez em 1876 por O. N. Witt para descrever grupos químicos associados à cor.
Microscópio de fluorescência [1][2][3]
[editar | editar código-fonte]As primeiras pesquisas que deram origem à invenção do microscópio de fluorescência atribuem-se ao cientista alemão August Köhler.
Köhler realizou experiências com radiação ultravioleta que, devido ao seu comprimento de onda mais curto, poderiam contribuir para o incremento do poder de resolução, face à já observada na microscopia óptica. Seria a partir destas pesquisas que surgiria o microscópio de ultravioleta (UV) (entre 1901 e 1904).
Em 1904, descobriu-se que o tecido demonstra fluorescência quando irradiado por luz ultravioleta, sendo este fenómeno conhecido como autofluorescência. Esta propriedade pesquisada na investigação de bactérias, protozoários, plantas, tecidos animais e em várias substâncias biorgânicas como, por exemplo, a albumina, a elastina e a queratina.
No século XX, nomeadamente em 4 de Abril de 1908, August Köhler e Henry Siedentopf, da companhia Carl Zeiss, apresentaram em um curso de microscopia dos primeiros microscópios de fluorescência, a partir do modelo de microscópio de UV.
Em 1914, Stanislav Von Prowazek utilizou o microscópio de fluorescência para estudar a ligação dos corantes às células vivas, chegando à conclusão que secções de tecido corados poderiam ser examinados recorrendo ao microscópio de fluorescência, caso os corantes utilizados fossem fluorescentes. Estes corantes foram denominados fluorocromos, de modo a distingui-los dos diacromos (tintas coloridas que se tornam visíveis por absorção de luz).
Contudo, o principal factor impulsionador para a microscopia de fluorescência deve-se ao desenvolvimento de técnicas de conjugação de fluorocromos a anticorpos por Coons e seus colaboradores, em 1941. A microscopia de fluorescência forneceu uma maior sensibilidade necessária para a demonstração das reacções anticorpo-antigénio in situ. A técnica de Coons marcava assim a génese da Imunofluorescência.
Com estes novos métodos, os microscópios de fluorescência tornaram-se acessíveis, levando à sua utilização em várias áreas.
A evolução tecnológica ligada ao microscópio de fluorescência assume carácter multidisciplinar uma vez que se cruza com a física, a óptica, a química, a engenharia genética e a biologia. Todas estas ciências deram um contributo fulcral para que fosse possível visualizar substâncias fluorescentes acopladas a proteínas e assim, aumentar a compreensão da actividade celular.
Cientistas como Wilhelm Conrad Roentgen, que venceu o primeiro Prémio Nobel da Física, e Alexandre Edmond Becquerel, fizeram as primeiras observações sobre a fluorescência. Antoine Henri Becquerel herdou do seu pai a colecção de minerais e compostos fluorescentes que, em associação com as conclusões da Roentgen, conseguiu demonstrar que tanto os raios-X como a fluorescência exigem uma contínua fonte de energia para emitir radiação.
A principal descoberta, que deu renome à microscopia de fluorescência, relaciona-se com uma proteína com autofluorescência de cor verde – proteína verde fluorescente (GFP, do inglês green fluorescent protein) – presente em organismos vivos. Foi identificada e isolada na década de 60 pelo biólogo marinho Osamu Shimomura a partir dos cnidários Aequorea victoria, sendo identificada como um brilho verde causado por uma proteína. Posteriormente, Douglas Prasher estudou o gene repórter da GFP. A estrutura desta proteína só foi devidamente caracterizada em 1996 por Martin Chalfie, Roger Y. Tsien e Osamu Shimomura.
Este conhecimento foi um marco importante para a evolução da área, promovendo uma nova classe de métodos de marcação de estruturas através da microscopia de fluorescência.
Um elemento-chave para a evolução da microscopia, em geral, foi a transição para a imagem digital, na década de 1990, com aquisição de instrumentos de alta sensibilidade e de computadores com hardware avançado, a preços acessíveis.
Os microscópios de fluorescência têm evoluído a uma velocidade espantosa ao longo da última década, aliada principalmente ao rápido avanço na tecnologia laser e do computador baseado em análise de imagem.
A microscopia de fluorescência é uma das técnicas de imagem mais utilizada para o estudo da dinâmica de células vivas, devido à sua capacidade de isolar diferentes proteínas com um alto grau de especificidade, sendo a sua sensibilidade suficientemente elevada para detectar cerca de 50 moléculas/µm3.
A marcação de diferentes moléculas com fluorocromos distintos (diferentes cores), permite a avaliação simultânea de vários tipos de moléculas. A combinação destes factores conferem à microscopia de fluorescência uma clara vantagem sobre outras técnicas observação estrutural, nomeadamente microscopia óptica, tanto in vitro como in vivo.
Através do desenvolvimento de sondas fluorescentes e novos microscópios de alta resolução, a biologia de imagem entrou numa nova era e tem, actualmente, um profundo impacto sobre a forma como a investigação é conduzida nas ciências da vida. Os cientistas têm vindo a depender cada vez mais da observação microscópica, sendo possível visualizar componentes e processos subcelulares in vivo, tanto ao nível estrutural como funcionalmente.
A importância da microscopia de fluorescência teve o seu reconhecimento máximo com a atribuição em 2008 do Prémio Nobel da Química, concedido aos cientistas que identificaram a estrutura da GFP.
A investigação de métodos de processamento de sinais biológicos é apenas o ponto de partida para a evolução desta área, sendo que ainda não estão claramente formuladas os campos a desenvolver.
No entanto, será previsível que os principais desafios na microscopia de fluorescência passem por métodos mais sensíveis e com maior amplificação de sinal.
A necessidade de resposta à investigação por parte da comunidade científica fomentará também a necessidade do fabrico de microscópios de fluorescência tecnologicamente mais avançados, assim como os acessórios adequados aos mesmos, o que constituirá uma importante contribuição para a pesquisa no âmbito dos organismos vivos.
Constituição do microscópio[7][8][9]
[editar | editar código-fonte]
Na microscopia de fluorescência, a amostra que se quer estudar é ela própria a fonte de luz, sendo a técnica usada para estudar amostras que possam emitir fluorescência.
O microscópio de fluorescência baseia-se no fenómeno que certo material emite energia detectável como luz visível quando irradiado com luz de um comprimento de onda específico. A amostra pode emitir fluorescência na sua forma natural, como a clorofila e alguns minerais, ou através de tratamento com químicos fluorescentes (fluoróforos).
O microscópio de fluorescência é constituído fundamentalmente por parte mecânica, que comporta o sistema de suporte o sistema de focagem, e por parte óptica, que comporta o sistema de ampliação e o sistema de iluminação.
Sistema de suporte[9][10]
[editar | editar código-fonte]O sistema de suporte serve para sustentar nas posições correctas as diferentes peças do microscópio e dele fazem parte o canhão, o revólver, o braço, a platina e a base do microscópio.
Este deve providenciar ao microscópio uma boa estabilidade mecânica, isto é, qualquer vibração entre a lâmina e o corpo do microscópio deve ser reduzida ao mínimo absoluto, uma vez que tal vibração pode ser aumentada pelo próprio factor de ampliação do microscópio.
A base e o braço do microscópio devem fornecer uma rígida estrutura de suporte para a platina e o corpo de forma a resistir às vibrações normais presentes num laboratório.
Os principais requisitos do sistema mecânico da platina são:
- Carga e descarga fácil (com o mínimo de manipulação ou dano)
- Movimentos ortogonais precisos nos eixos x e y
- Folga mínima nas engrenagens
- Necessidade de acoplamento entre os movimentos nas direcções x e y.
Além da estabilidade mecânica, o microscópio deve ter em conta os padrões de ergonomia estabelecidos para que o usuário desse equipamento se sinta confortável ao realizar a observação, principalmente se esta exigir maior dispêndio de tempo.
Sistema de focagem[9]
[editar | editar código-fonte]Este sistema serve para obter imagens nítidas do objecto que se está a observar.
Fazem parte deste sistema os parafusos macrométrico e micrométrico, sendo através deles que se procede à focagem.
Sistema de ampliação [9][10][11]
[editar | editar código-fonte]O sistema de ampliação serve para aumentar a imagem observada. A consecução deste objectivo é possível através das oculares e objectivas, que são sistemas de lentes convergentes.
Existem vários tipos de oculares devendo-se optar por aquela que mais se adequa ao trabalho pretendido. As de Huygens, por exemplo, têm como principais características a simplicidade de construção, baixo custo e desempenho adequado para muitas aplicações. Possuem uma cobertura de campo limitada e uma pequena tensão de relaxamento, que é a propriedade que a lente possui de evitar o cansaço da visão em observações muito longas.
Outros tipos de oculares são as Hi-Point, que oferecem a vantagem de tensão de relaxamento maior (bom para quem usa óculos) e possui a desvantagem de ter a cobertura de campo limitada. As Widefield oferecem maior cobertura de campo do que qualquer outra ocular e oferece uma tensão de relaxamento igual às Hi-Point. Já as Hyperplane Compensating evitam a aberração cromática lateral, isto é, distorções das cores na parte periférica do campo de observação, que é comum nas outras oculares e as Ultraplane são usadas especificamente para aplicações fotográficas.
No que diz respeito às objectivas, uma vez que a autofluorescência de iluminação de campo claro é perigosa para o ser vivo, as estas devem ser escolhidas cuidadosamente, sendo que em microscopia de fluorescência apenas as objectivas acromáticas simples são práticas.
Com iluminação de fundo escuro, o leque de objectivas passíveis de serem escolhidas é maior, sendo possível optar por lentes mais elaboradas, com maior abertura e maior “poder de recolha de luz”.
Sistema de iluminação[9]
[editar | editar código-fonte]O sistema de iluminação serve para fornecer a luz necessária à formação da imagem e consiste em: fonte de luz, filtros, condensador e diafragma de campo.
Este microscópio possui o sistema de iluminação de fluorescência situado acima do plano da amostra.
Fonte de luz[11]
[editar | editar código-fonte]Todas as fontes de luz emitem uma ampla faixa de comprimentos de onda, incluindo ultravioleta (UV) e luz visível azul, que são os comprimentos de onda de interesse na microscopia de fluorescência. Todavia, apenas algumas fontes de luz são adequadas, na medida em que emitem luz de ondas suficientemente baixas para uso prático.
Usualmente usam-se lâmpadas de gás a alta pressão, como as lâmpadas de vapor de mercúrio ou as lâmpadas de gás xénon. Para a excitação de alguns comprimentos de onda, na faixa da luz visível azul a verde, podem ainda ser usadas, por exemplo, lâmpadas com filamentos de halogéneo, pois produzem luz útil suficiente.
A escolha da fonte de luz adequada depende do tipo de trabalho realizado e para observações correntes é preferível usar-se as lâmpadas de mercúrio como fonte de luz, sendo que estas operam com corrente alternada e são mais baratas. Já as lâmpadas de xénon operam com corrente directa e requerem a adição de rectificadores ao equipamento primário, se usadas em alimentação normal. Com alimentação DC (corrente contínua) podem ser estabilizadas, sendo então adequadas para fluorimetria ou medição de emissão de fluorescência.
Ambos os tipos de lâmpadas diferem nas suas curvas de emissão. As lâmpadas de mercúrio, por exemplo, atingem amplitudes elevadas para determinados comprimentos de onda, enquanto que noutros a emissão é baixa, o que resulta num perfil de emissão com vários picos. As lâmpadas de xénon apresentam um perfil mais suave, com predominância de curvas contínuas. Fortuitamente, os picos na emissão por vapor de mercúrio coincidem com os comprimentos de onda de excitação de grande parte dos fluorocromos mais usados.
Tendo em conta que estas fontes de luz contém gás a elevada pressão, elas têm de ser manuseadas com grande cuidado e alojadas em estruturas fortes e com protecção, sendo que será aqui que se filtrarão o calor e emissão de infravermelhos antes da luz provinda da fonte iniciar o seu trajecto até à amostra.
Estas lâmpadas possuem ainda tempos de vida específicos, sendo, por exemplo, no caso das de mercúrio de aproximadamente 200 horas. Quando usadas para períodos de tempo superiores, pode resultar numa destruição da lâmpada com explosão desta e subsequente libertação do vapor de mercúrio nas imediações do microscópio.
Nos últimos anos, um novo tipo de fonte de luz tem sido introduzido, denominado de díodo de emissão de luz (LED). Este é um aparelho semicondutor de tipo estado sólido.
Os LEDs possuem períodos de vida muito longos, com poucas alterações na saída de luz ao longo do tempo de vida. Outra característica é que estas fontes de luz emitem num comprimento de onda apenas, com uma largura do pico muito estreita, o que faz com que o seu uso tenha aumentado ao nível da microscopia. Apesar de não serem tão intensas como as de alta pressão, estas não requerem tantos filtros ópticos para seleccionar o comprimento de onda de interesse e não sofrem as distorções de luz visualizadas nas de alta pressão. Além disto, não existe o perigo de explodirem.
Todavia, para emissão a diferentes comprimentos de onda, serão necessários diferentes LEDs, não sendo também necessário seleccionar um novo conjunto de filtros de emissão, pois cada LED emite em bandas muito estreitas e apenas num comprimento de onda.
Filtros[12]
[editar | editar código-fonte]Existem 3 grandes categorias de filtros: filtros de excitação (excitadores), filtros de barreira (emissão) e filtros separadores de raios dicromáticos ou espelhos dicromáticos.
Os filtros para fluorescência, inicialmente, foram construídos exclusivamente a partir de vidro corado ou uma “mistura” de gelatina entre dois vidros. Contudo, actualmente fabricam-se vidros de alta resolução com ópticas de interferência, nos filtros de excitação para que permitam ou rejeitem a passagem de luz a determinados comprimentos de onda com grande especificidade e elevada transmissão.
Os espelhos dicromáticos são filtros de interferência especializados, desenhados para reflectir ou permitir a passagem da luz a específicos comprimentos de onda, quando colocados no trajecto da luz com uma inclinação de 45º.
Os filtros de barreira são fabricados com ambos os vidros corados ou com revestimento de interferência (ou uma combinação dos dois).
Tipo de filtro |
Abreviaturas |
Significado
|
| Filtros de excitação | UG | Ultraviolet glass |
| BG | Blue glass | |
| KP ou SP | Shortpass filters | |
| IF | Interference filters: filtros bons em situações com baixo Stoke’s shift | |
| Filtros de barreira (se associado com número, este é referente ao comprimento de onda) | LP ou L | Longpass filters |
| Y ou GG | Yellow glass | |
| R ou RG | Red glass | |
| O ou OG | Orange glass | |
| K | Kante (termo alemão para bordo – filtro de bordo) | |
| BA | Filtro de barreira | |
| Filtros dicróicos separadores de feixes | CBS | Chromatic beam splitter |
| RKP | Reflection short pass |
Condensador [11]
[editar | editar código-fonte]Os condensadores de campo claro estão aptos a iluminar o objecto usando toda a energia disponível, no entanto, estes também direccionam os raios depois do objecto até a objectiva. Isto constitui, não só um potencial perigo para os olhos do observador, como pode também estabelecer uma perturbação na forma como os componentes da objectiva recebem a autofluorescência. Como consequência, a maioria dos sistemas adoptou o condensador de campo escuro que não permite a entrada de luz directa na objectiva, e adicionalmente, proporcionará um fundo escuro que contraste com a fluorescência.
A emissão de luz fluorescente é, na maioria dos casos, muito pobre relativamente à quantidade de energia absorvida pelos fluorocromos ou fluoróforos, com um ratio de eficácia compreendido entre 1:1000 e 1:100 (o melhor), de modo que qualquer sistema que reduza a energia disponível para qualquer medida deve ser bem considerada antes da sua utilização.
Diafragma [8]
[editar | editar código-fonte]O diafragma situa-se junto ao condensador e é o responsável pelo controlo da abertura angular do cone de luz para a iluminação da amostra.
Princípios físicos de funcionamento
[editar | editar código-fonte]Princípio geral [7]
[editar | editar código-fonte]
A microscopia de fluorescência assenta no seguinte fenómeno:
- Energia é absorvida pelo átomo, que fica excitado;
- O elétron, após absorver energia, orbita para um nível energético superior;
- O elétron retorna ao seu estado inicial emitindo um fóton.
Desta forma, a tarefa fulcral do microscópio de fluorescência consiste em permitir que a luz de excitação irradie a amostra e depois separar a luz emitida mais fraca para formar a imagem. Assim, primeiro, a luz, originária da fonte de luz colocada num extremo do microscópio, encontra-se frente a um filtro de excitação que apenas vai deixar passar a radiação com o comprimento de onda desejado, sendo este coincidente com o material fluorescente. A radiação depois passa pelo espelho dicromático, que faz com que rode 45º colidindo então com os átomos da amostra, o que leva a que os electrões sejam excitados para um nível energético superior. Quando estes átomos perdem a energia de excitação, retomam o nível energético de repouso e emitem luz (fotões).
Para se tornar visível, a luz emitida torna a passar pelo espelho dicromático sendo depois separada da luz de excitação (absorção) mais brilhante num filtro de barreira. Aqui, o facto de a luz emitida ter energia mais baixa e um maior comprimento de onda é usado para permitir a separação. As áreas fluorescentes são então exibidas e poder-se-ão observar no microscópio, evidenciadas contra um fundo escuro de alto contraste.
Desvio de Stokes [13]
[editar | editar código-fonte]
O desvio de Stokes corresponde à diferença (de comprimento de onda ou unidades de frequência) entre as posições no espectro dos pontos máximos das bandas de absorção e emissão de fluorescência para a mesma transição electrónica.
Geralmente, a emissão de fluorescência vai ocorrer a um comprimento de onda superior ao de absorção da mesma. Todavia, caso o fenómeno contrário se verifique é denominado por desvio anti-Stokes.
À medida que o desvio de Stokes aumenta, torna-se mais fácil separar luz de excitação da de emissão através do uso de combinações dos filtros.
Fenómenos de fading, quenching, photobleaching [12]
[editar | editar código-fonte]Existem um largo espectro de condições que regularmente surgem e afectam as radiações da emissão de fluorescência e vão reduzir a sua intensidade. O termo geral para a redução da intensidade de emissão de fluorescência é fading (desvanecimento), sendo este subdividido em fenómenos de quenching (extinção) e photobleaching (lixiviamento/branquemento).
O photobleaching corresponde à decomposição irreversível das moléculas fluorescentes no seu estado excitado devido à interacção destas moléculas com oxigénio molecular antes de emitirem energia. A ocorrência do photobleaching é explorada numa técnica conhecida por recuperação de fluorescência após photobleaching (FRAP – fluorescence recovery after photobleaching). O método baseia-se no branqueamento de uma região bem definida da amostra devido a uma intensa explosão de luz laser, acompanhada por uma subsequente observação das taxas e padrões da recuperação de fluorescência nas áreas branqueadas. Uma técnica relacionada, denominada perda de fluorescência em photobleaching (FLIP – fluorescence loss in photobleaching) é utilizada para monitorizar o decréscimo de fluorescência numa determinada região adjacente à área branqueada.
O processo de relaxamento após estado excitatório, no quenching, resulta numa redução da intensidade de fluorescência através de uma variedade de mecanismos que envolvem perda de energia não radioactiva e frequentemente ocorre como resultados de agentes oxidantes ou da presença de sais, metais pesados ou compostos de halogéneo. Em alguns casos, o quenching resulta da transmissão de energia para outra molécula (denominada aceitador) fisicamente próxima do fluoróforo excitado (o dador), sendo este fenómeno conhecido por ressonância de energia transferida em fluorescência (FRET – transferência de energia de ressonância por fluorescência).
Variação da luz fluorescente [12]
[editar | editar código-fonte]A emissão de fluorescência que resulta do fluxo de luz depende da absorção e emissão características do fluoróforo usado, da concentração deste na amostra e do comprimento do percurso óptico da amostra. Em termos matemáticos, a fluorescência produzida (F) é dada pela equação:
onde σ corresponde à absorção molecular do tecido, Q é o rendimento quântico, e I corresponde ao fluxo de luz incidente.
Partindo desta equação surge a questão de quantos fotões emitidos seriam detectados e por quanto tempo continuaria a taxa de emissão.
A eficiência de detecção é dada em função da eficiência do conjunto óptico usado e da eficiência do detector quântico.
Já a duração de emissão de fluorescência depende da taxa de destruição de fluoróforo como resultado do photobleaching. Uma forma de prolongar o período de observação é reduzindo a intensidade do fluxo de luz incidente, para que apenas uma pequena fracção das moléculas de fluoróforo na solução de coloração são excitadas e subsequentemente fotodestruídas.
Processamento de amostras [6][14][15]
[editar | editar código-fonte]A preparação de amostras para microscopia de fluorescência é, de uma forma geral, semelhante à preparação das amostras para os restantes tipos de microscópio. Contudo, existem alguns aspectos que devem ser tomados em consideração para a obtenção de bons resultados. De ressalvar que é importante evitar usar reagentes ou materiais que sejam fluorescentes ou que possam estar contaminados com materiais fluorescentes.
A técnica de preparação das amostras depende essencialmente do tipo de material. O material biológico é geralmente seccionado, em secções muito finas (na ordem das 3μm) que são colocadas em lâminas.
As amostras também podem ser utilizadas em forma de esfregaço (por exemplo de sangue, células esfoliadas, colónias de bactérias). Este tipo de material é colocado em lâmina e depois fixado.
Para além deste tipo de processamento, a microscopia de fluorescência também pode ser aplicada a células vivas, por exemplo, para determinar a concentração intracelular de iões de Ca2+ e H+, de forma a monitorizar o pH das células vivas.
O estudo em células vivas requer algumas condições especiais. Por exemplo para o estudo da fluorescência de NADH na camada superficial de tecidos vivos, é necessário manter a amostra numa caixa fechada com uma atmosfera controlada que previna a oxidação da NADH pelo oxigénio atmosférico.
Apesar de existirem múltiplas técnicas para a preparação das amostras, a mais utilizada consiste na colheita do tecido, fixação, processamento, inclusão, corte e coloração.
Fixação [6][16]
[editar | editar código-fonte]Após a sua remoção do organismo, os tecidos devem ser fixados o mais rapidamente possível, para evitar a digestão dos tecidos por enzimas presentes no interior das células (autólise) ou por bactérias (putrefacção), preservando assim a sua estrutura e a composição molecular.
A fixação poderá afectar a fluorescência do tecido. Fixadores como o formol e o ácido acético podem induzir a fluorescência em certas substâncias. O glutaraldeído produz fluorescência de fundo intensa.
Processamento [17]
[editar | editar código-fonte]O processamento é constituído pelas seguintes etapas:
- Desidratação – remoção de toda a água existente nos tecidos, através de sucessivos banhos de um agente desidratante. As soluções mais usadas são álcoois de concentração crescente.
- Diafanização ou clarificação – substituição do agente desidratante por uma substância que seja simultaneamente miscível em água e no meio de inclusão.
- Impregnação – preenchimento dos espaços tecidulares, que inicialmente continham água, por substância hidrofóba. Habitualmente, este produto será também o meio de inclusão.
- Inclusão
A inclusão coloca as amostras num meio de suporte, para que os tecidos adquiram uma consistência que permita a obtenção de cortes finos, que possam ser observados ao microscópio óptico.[17]
- Corte
Após a inclusão, as amostras em meio de suporte (blocos) são seccionadas num micrótomo de modo a obter cortes de 1-10 μm de espessura.[16]
- Marcação
Para seja possível o estudo dos cortes histológicos estes têm que ser corados, uma vez que, salvo raras excepções, a maioria dos tecidos é incolor.[16]
Para a microscopia de fluorescência são utilizados fluorocromos para observar as estruturas pretendidas. Os fluorocromos utilizados são moléculas fluorescentes com afinidade por moléculas presentes nas células que são utilizadas como corantes fluorescentes.[16]
- Montagem
A montagem assume especial importância na microscopia de fluorescência. O meio de montagem, uma vez que está em contacto com o fluorocromo, não poderá causar a sua dissolução ou obscurecimento.[6]
O meio de montagem para microscopia de fluorescência deve preencher alguns requisitos específicos. Assim sendo, este deve ser transparente a qualquer comprimento de onda, não ser fluorescente, o seu índice de refracção deve ser igual ao da lamela, não deve dissolver amostra em geral e o fluorocromo em particular, deve fornecer um ambiente propício ao fluorocromo, que maximize a sua fluorescência e evite reacções que levem ao seu enfraquecimento.[6]
Aplicações [11][18]
[editar | editar código-fonte]A técnica de microscopia de fluorescência tem-se tornado uma ferramenta essencial na biologia e nas ciências biomédicas bem como nas ciências dos materiais devido aos seus atributos.[3]
A utilização de fluorocromos tornou possível a identificação de células e de componentes celulares sub-microscópicos com um elevado grau de especificidade entre o material não fluorescente. De facto, o microscópio de fluorescência é capaz de revelar a presença de uma única molécula. Para além disso, este permite que através de técnicas de fluorescência múltipla possam ser identificadas, simultaneamente, várias moléculas alvo.
As proteínas fluorescentes são aplicadas para a determinação da localização e dinâmica de proteínas, organitos e outros compartimentos celulares, e até mesmo para avaliar a capacidade de expressão de diversos promotores. Para estes estudos são utilizadas amostras coradas com corantes fluorescentes (de uma forma geral, o isotiocianato de fluoresceína) ou amostras de substâncias que emitem luz de forma natural, quando expostas à luz ultravioleta ou azul.
As características deste microscópio, bem como a sua especificidade conferem às suas técnicas particular utilidade na exibição de estruturas e medição de eventos fisiológicos e bioquímicos nas células vivas. Particularmente, a existência de múltiplos indicadores fluorescentes contribui para o estudo de uma multiplicidade de produtos químicos fisiologicamente importantes como DNA, cálcio, magnésio, sódio, pH e enzimas.
Através da imunofluorescência, com recurso a anticorpos específicos, pode visualizar-se a presença das substâncias pretendidas, partindo da combinação de anticorpos específicos de várias moléculas biológicas, quimicamente ligados às moléculas fluorescentes, resultando na marcação de estruturas específicas dentro das células.
Deste modo, as aplicações da microscopia de fluorescência são várias permitindo âmbitos de estudo distintos. Alguns dos exemplos mais proeminentes são a pesquisa de organitos celulares, o estudo de reacções enzimáticas, análise de viabilidade celular e de função celular, como fenómenos de endocitose, exocitose, transdução de sinais, potenciais de membrana, entre outros.
FRAP [19][20]
[editar | editar código-fonte]No estudo funcional de macromoléculas, tais como de fenómenos de difusão, associação e dissociação, aplica-se um método denominado por FRAP (fluorescence recovery after photobleaching).
Este método consiste em projectar uma luz de grande intensidade para que um fluorocromo perca a capacidade de emitir fluorescência, podendo assim medir o movimento ao longo do tempo da molécula de interesse (por exemplo proteína, lípido ou hidrato de carbono), sendo que esta encontra-se covalentemente ligada a um outro fluorocromo, diferente do que sofreu photobleaching.
Esta técnica foi usada para ajudar a definir o modelo do mosaico fluido da membrana celular.
FRET [20][21]
[editar | editar código-fonte]A realização de experiências que envolvem moléculas adjacentes pode ser realizadas pelo método de FRET (Transferência de energia de ressonância por fluorescência).
Esta técnica permite demonstrar a distância entre uma molécula aceitadora e uma receptora devido à existência de ligações dipolo-dipolo.
Quando a molécula dadora se encontra a mais de 10 nm de distância da molécula receptora, haverá pouca interacção entre elas e a molécula dadora emitirá fotões após a sua excitação com o laser. No entanto, se as moléculas se encontrarem a uma distância inferior a 10 nm a molécula aceitadora receberá energia da molécula dadora e vai emitir fotões com uma cor diferente.
A eficácia da transferência de energia dependerá da distância entre o aceitador e o dador, do grau de sobreposição espectral entre o espectro de emissão do dador e o espectro de absorção do aceitador e da relativa orientação paralela dos dipolos do aceitador e do dador.
Também permite a realização de estudos de iões e moléculas metálicas ou sobre a distribuição e dinamismo intracelular de macromoléculas.
Vantagens e limitações
[editar | editar código-fonte]Vantagens[14][17]
[editar | editar código-fonte]- Permite o estudo e a observação de células vivas
- Como apenas a luz reflectida é observada, enquanto a transmitida é eliminada do campo de visão, cria maior intensidade e a definição da imagem é superior.
Limitações[22]
[editar | editar código-fonte]- Preparações não permanentes, a técnica de fluorescência vai decaindo, ou seja, ao fim de algum tempo de permanência do fluorocromo nas células, o seu efeito desaparece.
- Observação das preparações tem de ser efectuada em sala escura, devido à fraca adaptação do olho humano ao escuro, poderá ser difícil a observação de amostras durante algum tempo após o escurecimento da sala.
Galeria de imagens
[editar | editar código-fonte]-
 Imagem epifluorescente dos três componentes em uma divisão de uma célula cancerosa humana. O DNA está manchado de azul, uma proteína chamada INCENP aparece em verde, e os microtúbulos são vermelhos. Cada fluoróforo é processado separadamente utilizando uma combinação diferente de filtros de excitação e emissão, e as imagens são capturadas em sequência usando uma câmera CCD digital, em seguida, sobrepostas para dar uma imagem completa.
Imagem epifluorescente dos três componentes em uma divisão de uma célula cancerosa humana. O DNA está manchado de azul, uma proteína chamada INCENP aparece em verde, e os microtúbulos são vermelhos. Cada fluoróforo é processado separadamente utilizando uma combinação diferente de filtros de excitação e emissão, e as imagens são capturadas em sequência usando uma câmera CCD digital, em seguida, sobrepostas para dar uma imagem completa. -
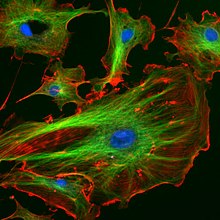 As células endoteliais sob o microscópio. Os núcleos são manchadis de azul com DAPI, os microtúbulos são marcados de verde por um anticorpo ligado ao FITC e filamentos de actina são rotulados em vermelho com a faloidina ligada ao TRITC. Células endoteliais da artéria pulmonar bovina (BPAE)
As células endoteliais sob o microscópio. Os núcleos são manchadis de azul com DAPI, os microtúbulos são marcados de verde por um anticorpo ligado ao FITC e filamentos de actina são rotulados em vermelho com a faloidina ligada ao TRITC. Células endoteliais da artéria pulmonar bovina (BPAE) -
 Microscopia 3D Dupla Cor de Super Resolução com Her2 e Her3 em células mamárias, corantes padrão: Alexa 488, Alexa 568. microscopia LIMON
Microscopia 3D Dupla Cor de Super Resolução com Her2 e Her3 em células mamárias, corantes padrão: Alexa 488, Alexa 568. microscopia LIMON -
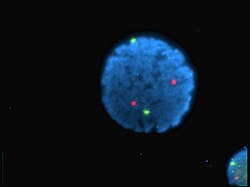 Núcleo de linfócitos humanos coloridos com DAPI com o cromossoma 13 (verde) e 21 (vermelho) sondas centrómero hibridizadas (hibridização fluorescente in situ (FISH))
Núcleo de linfócitos humanos coloridos com DAPI com o cromossoma 13 (verde) e 21 (vermelho) sondas centrómero hibridizadas (hibridização fluorescente in situ (FISH)) -
 Membrana da célula de levedura visualizada por algumas proteínas da membrana, fundidas com marcadores fluorescentes RFP e GFP. A imposição de luz de ambos os marcadores resulta na cor amarela.
Membrana da célula de levedura visualizada por algumas proteínas da membrana, fundidas com marcadores fluorescentes RFP e GFP. A imposição de luz de ambos os marcadores resulta na cor amarela. -
 Microscopia de Super Resolução: detecção de única molécula de YFP em uma célula cancerígena humana. Medidas típicas de distância na gama de 15 nm medido com um microscópio Vertico-SMI/SPDMphymod
Microscopia de Super Resolução: detecção de única molécula de YFP em uma célula cancerígena humana. Medidas típicas de distância na gama de 15 nm medido com um microscópio Vertico-SMI/SPDMphymod -
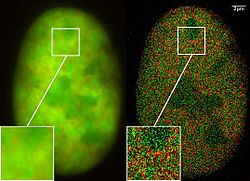 Microscopia de Super Resolução: Co-localização de microscopia (2CLM) com fusã de proeínas GFP e RFP (núcleo de uma célula de câncer ósseo) 120 000 moléculas localizadas em uma grande área (470 µm²) medido com um microcóspio Vertico-SMI/SPDMphymod
Microscopia de Super Resolução: Co-localização de microscopia (2CLM) com fusã de proeínas GFP e RFP (núcleo de uma célula de câncer ósseo) 120 000 moléculas localizadas em uma grande área (470 µm²) medido com um microcóspio Vertico-SMI/SPDMphymod -
 Microscopia de fluorescência de expressão de DNA no Human Wild-Type and P239S Mutant Palladin.
Microscopia de fluorescência de expressão de DNA no Human Wild-Type and P239S Mutant Palladin. -
 imagens de microscopia de fluorescência dA patologia flares solares em uma célula de sangue mostrando as áreas afetadas em vermelho.Microscópio de luz porem chamado também de microscopio óptico
imagens de microscopia de fluorescência dA patologia flares solares em uma célula de sangue mostrando as áreas afetadas em vermelho.Microscópio de luz porem chamado também de microscopio óptico









Referências
- ↑ a b Jameson, D. Principles of Fluorescence Techniques. 2009. Recuperado em 2009, Junho 4, de http://www.fluorescence-foundation.org/lectures/chicago2009/lecture1.pdf.
- ↑ a b c d e Jackson C. Fluorescence Microscopy and its Applications (sd). Recuperado em 2009, Maio 20, de http://www.andrew.cmu.edu/user/cjackso1/FluorescenceMicroscopy Applications.pdf
- ↑ a b «Visão de novos mundos». Ciência Hoje. Consultado em 21 de dezembro de 2020
- ↑ a b c d Vonesch C., Aguet F., Vonesch J.-L. & Unser M. The Colored Revolution of Bioimaging. IEEE Signal Processing Magazine. 2006; 23 (3): 20-31
- ↑ a b c d S/A. Technology Spotlight (2006). Recuperado em 2009, Maio 20 de http://www.biocompare.com/Articles/TechnologySpotlight/648/Fluorescent-Microscopy-True-Legends-For-Legendary-Views.html
- ↑ a b c d e f g h Rost, F. (1995) Fluorescence microscopy (Volume II). Sidney: Cambridge University Press
- ↑ a b The Fluorescence Microscope, 2009. Recuperado em 2009, Junho 29, de http://nobelprize.org/educational_games/physics/microscopes/fluorescence/index.html
- ↑ a b Cortez, T., Ferreira, A., Ferreira, S., Moreira, T. Constituição e Funcionamento do Microscópio Óptico Composto, 2002/2003. Recuperado em 2009, Julho 03, http://www.prof2000.pt/users/biologia/const_mic.htm
- ↑ a b c d e Microscópio, s.d.. Recuperado em 2009, Julho 03, de http://cienciacreazeitao.googlepages.com/microscopio.pdf
- ↑ a b Santos, E. Microscópio Óptico, s.d. Recuperado em 2009, Julho 03, de http://www.dsif.fee.unicamp.br/~furio/IE607A/MO.pdf
- ↑ a b c d Bancroft, J. D., Gamble, M. (2008) Theory and Practice of Histological Techniques (6ª Edição). UK: Churchill Livingstone
- ↑ a b c Davidson, M. W., & Spring, K. R. (s.d.). Nikon MicroscopyU - The source for microscopy education. Recuperado em 2009, Junho 29 , de http://www.microscopyu.com/articles/fluorescence/fluorescenceintro.html
- ↑ IUPAC Compendium of Chemical Terminology (2ª Edição), 1997. Recuperado em 2009, Junho 29, de http://www.iupac.org/goldbook/S06031.pdf}}
- ↑ a b Universidade Federal de Mato Grosso. Tipos de microscópio – microscópio de fluorescência comum. Recuperado em 2009, Junho 04, de http://www.ufmt.br/bionet/conteudos/01.09.04/fluor.htm
- ↑ Elisabete, A., Machado, J., Gonçalves, J., Oliveira, T. Técnicas de Imunohistoquímica de Fluorescência, 2009. Recuperado em 2009, Maio 30, de http://www.scribd.com/doc/14773321/Apresentacao-Tecnicas-de-Imunohistoquimica-de-Fluorescencia
- ↑ a b c d Junqueira, L., Carneiro, J. (2004) Histologia Básica (10ª edição). Rio de Janeiro: Guanabara Koogan
- ↑ a b c Moral, R. (1993) Laboratório de Anatomia Patológica (1ª edição). Madrid: McGraw-Hill
- ↑ Marchi, E. Revisão de Microscopia, 2005. Recuperado em 2009, Julho 02, de http://www.scribd.com/doc/6729026/Microscopia-basica2
- ↑ Campbell, M. Fluorescence Recovery After Photobleaching (FRAP), 2003. Recuperado em 2009, Junho 30, de http://www.bio.davidson.edu/Courses/Molbio/FRAPx/FRAP.html
- ↑ a b University of Illinois at Urbana-Champaign. Single-molecule fluorescence spectroscopy and microscopy, s.d. Recuperado em 2009, Junho 30, de https://netfiles.uiuc.edu/tjha/www/newTechnique.html
- ↑ Newbury, K. Fluorescence Resonance Energy Transfer, 2009. Recuperado em 2009, Junho 30, de http://www.icms.qmul.ac.uk/flowcytometry/uses/fret/index.html
- ↑ Davidson, M. Fluorescence Microscopy, 2009. Recuperado em 2009, Junho 4, de http://microscopy.fsu.edu/primer/techniques/fluorescence/fluorhome.html
Ligações externas
[editar | editar código-fonte]- «Biografia de August Köhler» (em inglês)
- «Preparação de espécimenes para microscopia de fluorescência» (em inglês)
- «Imagens de Microscopia de Fluorescência» (em inglês)
- «Simulador de Microscópio de fluorescência» (em inglês)